

What Is A Conjugated Protein
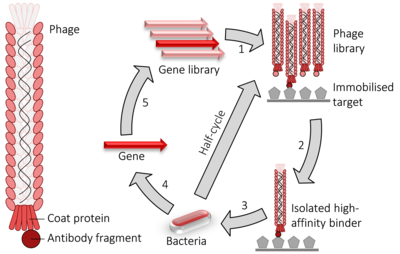
Phage brandish cycle. i) fusion proteins for a viral glaze protein + the gene to be evolved (typically an antibody fragment) are expressed in bacteriophage. 2) the library of phage are washed over an immobilised target. 3) the remaining loftier-affinity binders are used to infect bacteria. four) the genes encoding the high-affinity binders are isolated. 5) those genes may have random mutations introduced and used to perform another round of evolution. The pick and amplification steps can be performed multiple times at greater stringency to isolate higher-analogousness binders.
Phage display is a laboratory technique for the study of poly peptide–protein, poly peptide–peptide, and poly peptide–Deoxyribonucleic acid interactions that uses bacteriophages (viruses that infect bacteria) to connect proteins with the genetic information that encodes them.[1] In this technique, a gene encoding a protein of involvement is inserted into a phage coat poly peptide gene, causing the phage to "display" the protein on its outside while containing the gene for the protein on its inside, resulting in a connexion between genotype and phenotype. These displaying phages can so be screened confronting other proteins, peptides or DNA sequences, in order to notice interaction betwixt the displayed poly peptide and those other molecules. In this style, large libraries of proteins tin be screened and amplified in a process called in vitro option, which is analogous to natural choice.
The almost common bacteriophages used in phage display are M13 and fd filamentous phage,[2] [three] though T4,[4] T7, and λ phage accept besides been used.
History [edit]
Phage display was offset described past George P. Smith in 1985, when he demonstrated the display of peptides on filamentous phage (long, thin viruses that infect bacteria) past fusing the virus's capsid protein to one peptide out of a collection of peptide sequences.[ane] This displayed the different peptides on the outer surfaces of the collection of viral clones, where the screening pace of the process isolated the peptides with the highest binding affinity. In 1988, Stephen Parmley and George Smith described biopanning for analogousness choice and demonstrated that recursive rounds of pick could enrich for clones present at one in a billion or less.[five] In 1990, Jamie Scott and George Smith described creation of large random peptide libraries displayed on filamentous phage.[6] Phage display technology was farther developed and improved by groups at the Laboratory of Molecular Biology with Greg Wintertime and John McCafferty, The Scripps Inquiry Institute with Richard Lerner and Carlos Barbas and the German Cancer Enquiry Centre with Frank Breitling and Stefan Dübel for display of proteins such as antibodies for therapeutic protein engineering. Smith and Winter were awarded a one-half share of the 2018 Nobel Prize in chemical science for their contribution to developing phage display.[7] A patent by George Pieczenik claiming priority from 1985 also describes the generation of peptide libraries.[viii]
Principle [edit]
Similar the 2-hybrid organization, phage display is used for the high-throughput screening of protein interactions. In the example of M13 filamentous phage display, the Deoxyribonucleic acid encoding the protein or peptide of involvement is ligated into the pIII or pVIII gene, encoding either the minor or major coat protein, respectively. Multiple cloning sites are sometimes used to ensure that the fragments are inserted in all three possible reading frames so that the cDNA fragment is translated in the proper frame. The phage gene and insert Deoxyribonucleic acid hybrid is and then inserted (a procedure known every bit "transduction") into E. coli bacterial cells such as TG1, SS320, ER2738, or XL1-Blue East. coli. If a "phagemid" vector is used (a simplified display construct vector) phage particles will not be released from the E. coli cells until they are infected with helper phage, which enables packaging of the phage Dna and assembly of the mature virions with the relevant protein fragment as part of their outer coat on either the minor (pIII) or major (pVIII) coat protein. By immobilizing a relevant DNA or poly peptide target(s) to the surface of a microtiter plate well, a phage that displays a protein that binds to ane of those targets on its surface will remain while others are removed by washing. Those that remain can be eluted, used to produce more than phage (by bacterial infection with helper phage) and to produce a phage mixture that is enriched with relevant (i.due east. binding) phage. The repeated cycling of these steps is referred to as 'panning', in reference to the enrichment of a sample of gold past removing undesirable materials. Phage eluted in the last stride tin be used to infect a suitable bacterial host, from which the phagemids tin can be collected and the relevant Deoxyribonucleic acid sequence excised and sequenced to place the relevant, interacting proteins or poly peptide fragments.[ citation needed ]
The use of a helper phage tin can exist eliminated by using 'bacterial packaging cell line' engineering science.[9]
Elution can be done combining depression-pH elution buffer with sonification, which, in add-on to loosening the peptide-target interaction, also serves to detach the target molecule from the immobilization surface. This ultrasound-based method enables single-step selection of a high-affinity peptide.[10]
Applications [edit]
Applications of phage display applied science include determination of interaction partners of a protein (which would be used equally the immobilised phage "bait" with a DNA library consisting of all coding sequences of a jail cell, tissue or organism) so that the function or the mechanism of the function of that protein may be adamant.[eleven] Phage display is as well a widely used method for in vitro poly peptide evolution (also called protein applied science). As such, phage display is a useful tool in drug discovery. It is used for finding new ligands (enzyme inhibitors, receptor agonists and antagonists) to target proteins.[12] [13] [xiv] The technique is also used to determine tumour antigens (for use in diagnosis and therapeutic targeting)[15] and in searching for protein-Dna interactions[16] using specially-constructed DNA libraries with randomised segments. Recently, phage display has also been used in the context of cancer treatments - such as the adoptive cell transfer arroyo.[17] In these cases, phage display is used to create and select synthetic antibodies that target tumour surface proteins.[17] These are made into synthetic receptors for T-Cells collected from the patient that are used to combat the disease.[18]
Competing methods for in vitro protein evolution include yeast brandish, bacterial display, ribosome display, and mRNA display.[ citation needed ]
Antibody maturation in vitro [edit]
The invention of antibody phage display revolutionised antibiotic drug discovery. Initial piece of work was washed past laboratories at the MRC Laboratory of Molecular Biology (Greg Winter and John McCafferty), the Scripps Research Institute (Richard Lerner and Carlos F. Barbas) and the German Cancer Research Centre (Frank Breitling and Stefan Dübel).[19] [20] [21] In 1991, The Scripps group reported the starting time brandish and selection of human antibodies on phage.[22] This initial study described the rapid isolation of human antibody Fab that bound tetanus toxin and the method was and then extended to rapidly clone human anti-HIV-1 antibodies for vaccine design and therapy.[23] [24] [25] [26] [27]
Phage display of antibody libraries has become a powerful method for both studying the immune response besides as a method to rapidly select and evolve human antibodies for therapy. Antibiotic phage display was later used past Carlos F. Barbas at The Scripps Research Establish to create synthetic human antibody libraries, a principle first patented in 1990 by Breitling and coworkers (Patent CA 2035384), thereby allowing human antibodies to be created in vitro from synthetic diverseness elements.[28] [29] [30] [31]
Antibody libraries displaying millions of unlike antibodies on phage are often used in the pharmaceutical manufacture to isolate highly specific therapeutic antibiotic leads, for development into antibody drugs primarily as anti-cancer or anti-inflammatory therapeutics. One of the most successful was adalimumab, discovered by Cambridge Antibody Engineering science equally D2E7 and developed and marketed past Abbott Laboratories. Adalimumab, an antibiotic to TNF alpha, was the earth's first fully human antibody[32] to achieve almanac sales exceeding $1bn.[33]
Full general protocol [edit]
Below is the sequence of events that are followed in phage display screening to identify polypeptides that bind with high affinity to desired target protein or DNA sequence:[ commendation needed ]
- Target proteins or Deoxyribonucleic acid sequences are immobilized to the wells of a microtiter plate.
- Many genetic sequences are expressed in a bacteriophage library in the form of fusions with the bacteriophage glaze protein, and so that they are displayed on the surface of the viral particle. The protein displayed corresponds to the genetic sequence within the phage.
- This phage-brandish library is added to the dish and later on assuasive the phage time to bind, the dish is done.
- Phage-displaying proteins that collaborate with the target molecules remain attached to the dish, while all others are washed abroad.
- Attached phage may be eluted and used to create more than phage by infection of suitable bacterial hosts. The new phage constitutes an enriched mixture, containing considerably less irrelevant phage (i.e. non-bounden) than were nowadays in the initial mixture.
- Steps 3 to 5 are optionally repeated ane or more times, further enriching the phage library in binding proteins.
- Following farther bacterial-based distension, the DNA within in the interacting phage is sequenced to identify the interacting proteins or protein fragments.
Selection of the coat protein [edit]
Filamentous phages [edit]
pIII [edit]
pIII is the protein that determines the infectivity of the virion. pIII is composed of three domains (N1, N2 and CT) continued by glycine-rich linkers.[34] The N2 domain binds to the F hair during virion infection freeing the N1 domain which so interacts with a TolA poly peptide on the surface of the bacterium.[34] Insertions within this protein are usually added in position 249 (within a linker region between CT and N2), position 198 (within the N2 domain) and at the Due north-terminus (inserted between the N-terminal secretion sequence and the N-terminus of pIII).[34] Even so, when using the BamHI site located at position 198 one must be careful of the unpaired Cysteine residue (C201) that could cause problems during phage brandish if one is using a non-truncated version of pIII.[34]
An reward of using pIII rather than pVIII is that pIII allows for monovalent brandish when using a phagemid (plasmid derived from Ff phages) combined with a helper phage. Moreover, pIII allows for the insertion of larger protein sequences (>100 amino acids)[35] and is more than tolerant to information technology than pVIII. Yet, using pIII as the fusion partner can lead to a decrease in phage infectivity leading to problems such as choice bias caused past difference in phage growth charge per unit[36] or even worse, the phage'due south inability to infect its host.[34] Loss of phage infectivity can be avoided by using a phagemid plasmid and a helper phage and then that the resultant phage contains both wild type and fusion pIII.[34]
cDNA has also been analyzed using pIII via a two complementary leucine zippers organisation,[37] Directly Interaction Rescue[38] or by adding an 8-10 amino acid linker between the cDNA and pIII at the C-terminus.[39]
pVIII [edit]
pVIII is the master coat protein of Ff phages. Peptides are usually fused to the N-terminus of pVIII.[34] Usually peptides that can exist fused to pVIII are 6-viii amino acids long.[34] The size brake seems to accept less to practise with structural impediment caused past the added department[xl] and more to exercise with the size exclusion acquired by pIV during glaze protein export.[40] Since there are effectually 2700 copies of the poly peptide on a typical phages, information technology is more probable that the protein of interest will be expressed polyvalently even if a phagemid is used.[34] This makes the apply of this protein unfavorable for the discovery of high analogousness binding partners.[34]
To overcome the size problem of pVIII, artificial glaze proteins accept been designed.[41] An case is Weiss and Sidhu's inverted bogus coat protein (ACP) which allows the brandish of big proteins at the C-terminus.[41] The ACP'south could display a protein of 20kDa, however, but at low levels (mostly but monovalently).[41]
pVI [edit]
pVI has been widely used for the brandish of cDNA libraries.[34] The display of cDNA libraries via phage display is an attractive alternative to the yeast-ii-hybrid method for the discovery of interacting proteins and peptides due to its high throughput capability.[34] pVI has been used preferentially to pVIII and pIII for the expression of cDNA libraries because one tin add the protein of interest to the C-terminus of pVI without greatly affecting pVI's function in phage assembly. This means that the end codon in the cDNA is no longer an issue.[42] All the same, phage display of cDNA is ever express by the inability of most prokaryotes in producing post-translational modifications present in eukaryotic cells or by the misfolding of multi-domain proteins.
While pVI has been useful for the analysis of cDNA libraries, pIII and pVIII remain the nearly utilized coat proteins for phage display.[34]
pVII and pIX [edit]
In an experiment in 1995, display of Glutathione Southward-transferase was attempted on both pVII and pIX and failed.[43] Still, phage display of this protein was completed successfully after the addition of a periplasmic signal sequence (pelB or ompA) on the N-terminus.[44] In a contempo report, it has been shown that AviTag, FLAG and His could be displayed on pVII without the need of a signal sequence. Then the expression of unmarried chain Fv'due south (scFv), and single chain T cell receptors (scTCR) were expressed both with and without the indicate sequence.[45]
PelB (an amino acrid bespeak sequence that targets the poly peptide to the periplasm where a signal peptidase and then cleaves off PelB) improved the phage display level when compared to pVII and pIX fusions without the indicate sequence. However, this led to the incorporation of more than helper phage genomes rather than phagemid genomes. In all cases, phage display levels were lower than using pIII fusion. Yet, lower display might be more than favorable for the selection of binders due to lower display being closer to truthful monovalent brandish. In 5 out of six occasions, pVII and pIX fusions without pelB was more efficient than pIII fusions in affinity option assays. The paper even goes on to country that pVII and pIX display platforms may outperform pIII in the long run.[45]
The use of pVII and pIX instead of pIII might too be an reward considering virion rescue may be undertaken without breaking the virion-antigen bond if the pIII used is wild type. Instead, one could cleave in a section betwixt the dewdrop and the antigen to elute. Since the pIII is intact information technology does not matter whether the antigen remains bound to the phage.[45]
T7 phages [edit]
The issue of using Ff phages for phage display is that they require the protein of interest to be translocated across the bacterial inner membrane before they are assembled into the phage.[46] Some proteins cannot undergo this process and therefore cannot be displayed on the surface of Ff phages. In these cases, T7 phage display is used instead.[46] In T7 phage display, the protein to exist displayed is attached to the C-terminus of the gene ten capsid protein of T7.[46]
The disadvantage of using T7 is that the size of the protein that tin be expressed on the surface is limited to shorter peptides because big changes to the T7 genome cannot be accommodated like it is in M13 where the phage merely makes its coat longer to fit the larger genome within information technology. Notwithstanding, information technology can be useful for the production of a big poly peptide library for scFV choice where the scFV is expressed on an M13 phage and the antigens are expressed on the surface of the T7 phage.[47]
Bioinformatics resources and tools [edit]
Databases and computational tools for mimotopes take been an important part of phage display study.[48] Databases,[49] programs and spider web servers[50] have been widely used to exclude target-unrelated peptides,[51] characterize small molecules-protein interactions and map protein-protein interactions. Users can use three dimensional structure of a protein and the peptides selected from phage brandish experiment to map conformational epitopes. Some of the fast and efficient computational methods are bachelor online.[50]
See also [edit]
- Directed evolution
- protein–protein interactions
- PelB leader sequence
Competing techniques:
- Two-hybrid arrangement
- mRNA display
- Ribosome display
References [edit]
- ^ a b Smith GP (June 1985). "Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface". Science. 228 (4705): 1315–7. Bibcode:1985Sci...228.1315S. doi:10.1126/science.4001944. PMID 4001944.
- ^ Smith GP, Petrenko VA (April 1997). "Phage Brandish". Chem. Rev. 97 (2): 391–410. doi:10.1021/cr960065d. PMID 11848876.
- ^ Kehoe JW, Kay BK (November 2005). "Filamentous phage display in the new millennium". Chem. Rev. 105 (11): 4056–72. doi:x.1021/cr000261r. PMID 16277371.
- ^ Malys N, Chang DY, Baumann RG, Xie D, Black LW (2002). "A bipartite bacteriophage T4 SOC and HOC randomized peptide display library: detection and analysis of phage T4 terminase (gp17) and late sigma factor (gp55) interaction". J Mol Biol. 319 (ii): 289–304. doi:10.1016/S0022-2836(02)00298-X. PMID 12051907.
- ^ Parmley SF, Smith GP (1988). "Antibody-selectable filamentous fd phage vectors: affinity purification of target genes". Gene. 73 (two): 305–318. doi:10.1016/0378-1119(88)90495-7. PMID 3149606.
- ^ Scott, J.; Smith, G. (1990). "Searching for peptide ligands with an epitope library". Science. 249 (4967): 386–390. Bibcode:1990Sci...249..386S. doi:x.1126/science.1696028. PMID 1696028.
- ^ "The Nobel Prize in Chemistry 2018". NobelPrize.org . Retrieved 2018-10-03 .
- ^ Usa patent 5866363, Pieczenik Chiliad, "Method and means for sorting and identifying biological information", published 1999-02-02
- ^ Chasteen L, Ayriss J, Pavlik P, Bradbury AR (2006). "Eliminating helper phage from phage brandish". Nucleic Acids Res. 34 (21): e145. doi:10.1093/nar/gkl772. PMC1693883. PMID 17088290.
- ^ Lunder Yard, Bratkovic T, Urleb U, Kreft S, Strukelj B (June 2008). "Ultrasound in phage brandish: a new approach to nonspecific elution". BioTechniques. 44 (7): 893–900. doi:10.2144/000112759. PMID 18533899.
- ^ Caption of "Protein interaction mapping" from The Wellcome Trust
- ^ Lunder M, Bratkovic T, Doljak B, Kreft S, Urleb U, Strukelj B, Plazar N (November 2005). "Comparing of bacterial and phage brandish peptide libraries in search of target-binding motif". Appl. Biochem. Biotechnol. 127 (ii): 125–31. doi:10.1385/ABAB:127:ii:125. PMID 16258189. S2CID 45243314.
- ^ Bratkovic T, Lunder M, Popovic T, Kreft Southward, Turk B, Strukelj B, Urleb U (July 2005). "Affinity selection to papain yields stiff peptide inhibitors of cathepsins L, B, H, and Grand". Biochem. Biophys. Res. Commun. 332 (3): 897–903. doi:ten.1016/j.bbrc.2005.05.028. PMID 15913550.
- ^ Lunder M, Bratkovic T, Kreft S, Strukelj B (July 2005). "Peptide inhibitor of pancreatic lipase selected past phage display using dissimilar elution strategies". J. Lipid Res. 46 (7): 1512–6. doi:ten.1194/jlr.M500048-JLR200. PMID 15863836.
- ^ Hufton SE, Moerkerk PT, Meulemans EV, de Bruïne A, Arends JW, Hoogenboom 60 minutes (December 1999). "Phage display of cDNA repertoires: the pVI brandish system and its applications for the choice of immunogenic ligands". J. Immunol. Methods. 231 (ane–2): 39–51. doi:10.1016/S0022-1759(99)00139-8. PMID 10648926.
- ^ Gommans WM, Haisma HJ, Rots MG (December 2005). "Applied science zinc finger protein transcription factors: the therapeutic relevance of switching endogenous cistron expression on or off at command". J. Mol. Biol. 354 (three): 507–19. doi:x.1016/j.jmb.2005.06.082. PMID 16253273.
- ^ a b "Automobile T Cells: Technology Patients' Immune Cells to Care for Their Cancers". National Cancer Institute. 2013-12-06. Retrieved 9 February 2018.
- ^ Løset GÅ, Berntzen 1000, Frigstad T, Pollmann S, Gunnarsen KS, Sandlie I (12 January 2015). "Phage Display Engineered T Cell Receptors as Tools for the Written report of Tumor Peptide-MHC Interactions". Frontiers in Oncology. 4 (378): 378. doi:10.3389/fonc.2014.00378. PMC4290511. PMID 25629004.
- ^ McCafferty J, Griffiths AD, Wintertime G, Chiswell DJ (December 1990). "Phage antibodies: filamentous phage displaying antibody variable domains". Nature. 348 (6301): 552–4. Bibcode:1990Natur.348..552M. doi:10.1038/348552a0. PMID 2247164. S2CID 4258014.
- ^ Scott JS, Barbas CF Iii, Burton DA (2001). Phage Display: A Laboratory Manual. Plainview, N.Y: Common cold Spring Harbor Laboratory Press. ISBN978-0-87969-740-2.
- ^ Breitling F, Dübel Southward, Seehaus T, Klewinghaus I, Piddling M (August 1991). "A surface expression vector for antibody screening". Gene. 104 (2): 147–53. doi:10.1016/0378-1119(91)90244-six. PMID 1916287.
- ^ Barbas CF, Kang Equally, Lerner RA, Benkovic SJ (September 1991). "Assembly of combinatorial antibody libraries on phage surfaces: the cistron III site". Proceedings of the National Academy of Sciences of the United states of america. 88 (eighteen): 7978–82. Bibcode:1991PNAS...88.7978B. doi:10.1073/pnas.88.18.7978. PMC52428. PMID 1896445.
- ^
- ^ Barbas CF, Björling E, Chiodi F, Dunlop N, Cababa D, Jones TM, Zebedee SL, Persson MA, Nara PL, Norrby E (October 1992). "Recombinant man Fab fragments neutralize human type 1 immunodeficiency virus in vitro". Proceedings of the National Academy of Sciences of the United States of America. 89 (xix): 9339–43. Bibcode:1992PNAS...89.9339B. doi:10.1073/pnas.89.19.9339. PMC50122. PMID 1384050.
- ^ Burton DR, Pyati J, Koduri R, Abrupt SJ, Thornton GB, Parren Pow, Sawyer LS, Hendry RM, Dunlop N, Nara PL (November 1994). "Efficient neutralization of primary isolates of HIV-1 past a recombinant human being monoclonal antibody". Science. 266 (5187): 1024–7. Bibcode:1994Sci...266.1024B. doi:ten.1126/scientific discipline.7973652. PMID 7973652.
- ^ Yang WP, Green Thousand, Pinz-Sweeney Southward, Briones AT, Burton DR, Barbas CF (December 1995). "CDR walking mutagenesis for the affinity maturation of a potent human anti-HIV-one antibody into the picomolar range". Periodical of Molecular Biology. 254 (3): 392–403. doi:x.1006/jmbi.1995.0626. PMID 7490758.
- ^ Barbas CF, Hu D, Dunlop Northward, Sawyer 50, Cababa D, Hendry RM, Nara PL, Burton DR (April 1994). "In vitro evolution of a neutralizing human antibody to homo immunodeficiency virus type one to enhance analogousness and augment strain cross-reactivity". Proceedings of the National Academy of Sciences of the United states of America. 91 (9): 3809–thirteen. Bibcode:1994PNAS...91.3809B. doi:ten.1073/pnas.91.9.3809. PMC43671. PMID 8170992.
- ^ Barbas CF, Bain JD, Hoekstra DM, Lerner RA (May 1992). "Semisynthetic combinatorial antibody libraries: a chemical solution to the multifariousness problem". Proc. Natl. Acad. Sci. U.s.A. 89 (x): 4457–61. Bibcode:1992PNAS...89.4457B. doi:10.1073/pnas.89.10.4457. PMC49101. PMID 1584777.
- ^ Barbas CF, Languino LR, Smith JW (November 1993). "High-affinity self-reactive human being antibodies by design and pick: targeting the integrin ligand bounden site". Proc. Natl. Acad. Sci. U.S.A. 90 (21): 10003–7. Bibcode:1993PNAS...9010003B. doi:10.1073/pnas.xc.21.10003. PMC47701. PMID 7694276.
- ^ Barbas CF, Wagner J (Oct 1995). "Synthetic Man Antibodies: Selecting and Evolving Functional Proteins". Methods. 8 (2): 94–103. doi:ten.1006/meth.1995.9997.
- ^ Barbas CF (Baronial 1995). "Synthetic human antibodies". Nat. Med. one (8): 837–9. doi:10.1038/nm0895-837. PMID 7585190. S2CID 6983649.
- ^ Lawrence Due south (Apr 2007). "Billion dollar babies--biotech drugs equally blockbusters". Nat. Biotechnol. 25 (4): 380–two. doi:10.1038/nbt0407-380. PMID 17420735. S2CID 205266758.
- ^ Cambridge Antibiotic: Sales update | Company Announcements | Telegraph
- ^ a b c d e f g h i j one thousand l g Lowman HB, Clackson T (2004). "i.iii". Phage brandish: a practical approach. Oxford [Oxfordshire]: Oxford University Press. pp. 10–eleven. ISBN978-0-19-963873-4.
- ^ Sidhu SS, Weiss GA, Wells JA (February 2000). "Loftier copy display of large proteins on phage for functional selections". J. Mol. Biol. 296 (2): 487–95. doi:10.1006/jmbi.1999.3465. PMID 10669603.
- ^ Derda R, Tang SK, Whitesides GM (July 2010). "Uniform amplification of phage with different growth characteristics in private compartments consisting of monodisperse aerosol". Angew. Chem. Int. Ed. Engl. 49 (31): 5301–4. doi:ten.1002/anie.201001143. PMC2963104. PMID 20583018.
- ^ Crameri R, Jaussi R, Menz Yard, Blaser K (Nov 1994). "Brandish of expression products of cDNA libraries on phage surfaces. A versatile screening system for selective isolation of genes by specific cistron-product/ligand interaction". Eur. J. Biochem. 226 (1): 53–viii. doi:10.1111/j.1432-1033.1994.tb20025.x. PMID 7957259.
- ^ Gramatikoff K, Georgiev O, Schaffner W (December 1994). "Direct interaction rescue, a novel filamentous phage technique to study protein-poly peptide interactions". Nucleic Acids Res. 22 (25): 5761–ii. doi:10.1093/nar/22.25.5761. PMC310144. PMID 7838733.
- ^ Fuh Grand, Sidhu SS (September 2000). "Efficient phage display of polypeptides fused to the carboxy-terminus of the M13 factor-3 small coat protein". FEBS Lett. 480 (2–3): 231–4. doi:10.1016/s0014-5793(00)01946-three. PMID 11034335. S2CID 23009887.
- ^ a b Malik P, Terry TD, Bellintani F, Perham RN (October 1998). "Factors limiting display of foreign peptides on the major glaze protein of filamentous bacteriophage capsids and a potential part for leader peptidase". FEBS Lett. 436 (2): 263–6. doi:10.1016/s0014-5793(98)01140-5. PMID 9781692. S2CID 19331069.
- ^ a b c Weiss GA, Sidhu SS (June 2000). "Pattern and evolution of artificial M13 coat proteins". J. Mol. Biol. 300 (1): 213–9. doi:10.1006/jmbi.2000.3845. PMID 10864510.
- ^ Jespers LS, Messens JH, De Keyser A, Eeckhout D, Van den Brande I, Gansemans YG, Lauwereys MJ, Vlasuk GP, Stanssens PE (April 1995). "Surface expression and ligand-based selection of cDNAs fused to filamentous phage gene VI". Bio/Engineering. 13 (4): 378–82. doi:10.1038/nbt0495-378. PMID 9634780. S2CID 6171262.
- ^ Endemann H, Model P (July 1995). "Location of filamentous phage minor coat proteins in phage and in infected cells". J. Mol. Biol. 250 (4): 496–506. doi:ten.1006/jmbi.1995.0393. PMID 7616570.
- ^ Gao C, Mao S, Lo CH, Wirsching P, Lerner RA, Janda KD (May 1999). "Making artificial antibodies: a format for phage display of combinatorial heterodimeric arrays". Proc. Natl. Acad. Sci. U.South.A. 96 (eleven): 6025–30. Bibcode:1999PNAS...96.6025G. doi:10.1073/pnas.96.eleven.6025. PMC26829. PMID 10339535.
- ^ a b c Løset GÅ, Roos Due north, Bogen B, Sandlie I (2011). "Expanding the versatility of phage display II: improved affinity selection of folded domains on poly peptide Vii and IX of the filamentous phage". PLOS One. 6 (2): e17433. Bibcode:2011PLoSO...617433L. doi:10.1371/journal.pone.0017433. PMC3044770. PMID 21390283.
- ^ a b c Danner S, Belasco JG (November 2001). "T7 phage display: a novel genetic option system for cloning RNA-binding proteins from cDNA libraries". Proc. Natl. Acad. Sci. U.S.A. 98 (23): 12954–nine. Bibcode:2001PNAS...9812954D. doi:x.1073/pnas.211439598. PMC60806. PMID 11606722.
- ^ Castillo J, Goodson B, Wintertime J (November 2001). "T7 displayed peptides as targets for selecting peptide specific scFvs from M13 scFv display libraries". J. Immunol. Methods. 257 (ane–2): 117–22. doi:10.1016/s0022-1759(01)00454-nine. PMID 11687245.
- ^ Huang J, Ru B, Dai P (2011). "Bioinformatics resources and tools for phage display". Molecules. xvi (1): 694–709. doi:ten.3390/molecules16010694. PMC6259106. PMID 21245805.
- ^ Huang J, Ru B, Zhu P, Nie F, Yang J, Wang Ten, Dai P, Lin H, Guo FB, Rao N (January 2012). "MimoDB ii.0: a mimotope database and beyond". Nucleic Acids Res. 40 (Database issue): D271–7. doi:10.1093/nar/gkr922. PMC3245166. PMID 22053087.
- ^ a b Negi SS, Braun Due west (2009). "Automated Detection of Conformational Epitopes Using Phage Display Peptide Sequences". Bioinform Biol Insights. 3: 71–81. doi:10.4137/BBI.S2745. PMC2808184. PMID 20140073.
- ^ Huang J, Ru B, Li Southward, Lin H, Guo FB (2010). "SAROTUP: scanner and reporter of target-unrelated peptides". J. Biomed. Biotechnol. 2010: 101932. doi:10.1155/2010/101932. PMC2842971. PMID 20339521.
Further reading [edit]
- Ledsgaard Fifty, Kilstrup Yard, Karatt-Vellatt A, McCafferty J, Laustsen AH (2018). "Basics of antibody phage display technology" (PDF). Toxins. 10 (6): 236. doi:x.3390/toxins10060236. PMC6024766. PMID 29890762.
- Selection Versus Pattern in Chemic Technology
- The ETH-2 human antibiotic phage library
- Sidhu SS, Lowman HB, Cunningham BC, Wells JA (2000). "Phage display for pick of novel bounden peptides". Meth. Enzymol. Methods in Enzymology. 328: 333–63. doi:10.1016/S0076-6879(00)28406-1. ISBN9780121822293. PMID 11075354.
External links [edit]

What Is A Conjugated Protein,
Source: https://en.wikipedia.org/wiki/Phage_display
Posted by: ruizlacky1937.blogspot.com
0 Response to "What Is A Conjugated Protein"
Post a Comment